


MDR a wyroby medyczne- Czy jesteś przygotowany na zmiany? Część I: Producent
Jakie są nowe obowiązki prawne dla producentów wyrobów medycznych?
Czy w Twoim portfolio znajdują się wyroby medyczne klasy I i planujesz ich sprzedaż również po 2024 roku?
Chcesz rozwijać innowacyjne wyroby medyczne „farmaceutyczne” zgodnie z nowym rozporządzeniem MDR?
26 maja 2021 roku wchodzą w życie ważne zmiany przepisów UE w zakresie wyrobów medycznych. Przepisy reguluje głownie ROZPORZĄDZENIE PARLAMENTU EUROPEJSKIEGO I RADY (UE) 2017/745 z dnia 5 kwietnia 2017 r. (Rozporządzenie MDR) w sprawie wyrobów medycznych.
Główne cele Rozporządzenia MDR, które zastąpi dotychczasowe dyrektywy unijne, to ustanowienie jednolitych regulacji dla rynku wyrobów medycznych w Unii Europejskiej, łatwiejsza identyfikacja samych wyrobów oraz podmiotów odpowiedzialnych za ich wytwarzanie i dystrybucję.
Nowe przepisy nakładają szereg obowiązków dla wytwórców oraz dystrybutorów wyrobów medycznych. Celem niniejszego artykułu jest prezentacja najważniejszych zmian dla producenta wyrobów medycznych z perspektywy firmy prowadzącej rozwój oraz sprzedaż wyrobów medycznych.
Jeśli chcesz dowiedzieć się jakie są nowe obowiązki dystrybutorów – kliknij w TEN link.
MDR a wyroby medyczne – harmonogram zmian
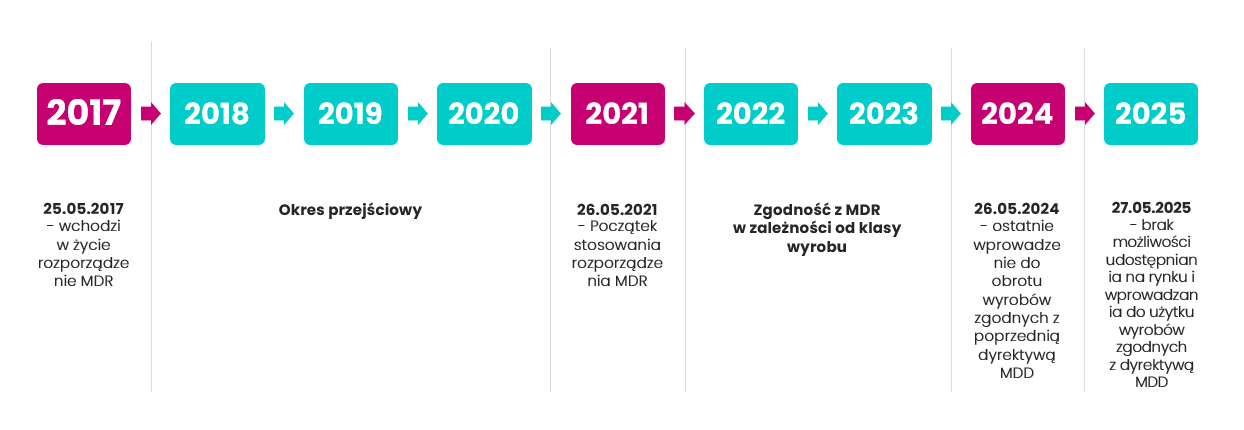
Jak wynika z powyższej grafiki, najważniejsze daty dla producentów wyrobów medycznych to:
– 26 maja 2021: obowiązują nowe przepisy dla wyrobów medycznych zgodnie z MDR,
–26 maja 2024: do tej daty możesz wprowadzać do obrotu wyroby medyczne zgodnie jeszcze z Dyrektywą MDD (np. w klasie I, dla których została przeprowadzona ocena zgodności i wystawiona deklaracja zgodności przed majem 2021),
– 27 maja 2025 r. to data, do której Twoje produkty zgodne z MDD mogą pozostawać na rynku w obrocie. Po tym terminie nie mogą być dłużej sprzedawane
Najważniejsze zmiany wprowadzone przez MDR dla producenta wyrobów medycznych
Portfolio produktowe
Czy switch do wyższej klasy dotyczy wszystkich wyrobów medycznych?
- należy dokonać przeglądu aktualnego portfolio produktowego pod kątem nowych wymagań MDR, aby określić konieczne działania.
- rozporządzenie wnosi zmianę w zakresie reguł klasyfikacji, np. dla wyrobów medycznych składających się z substancji lub mieszanin substancji, które są przeznaczone do wprowadzenia do organizmu ludzkiego przez jeden z otworów ciała lub do podania na skórę i które są wchłaniane przez organizm ludzki lub miejscowo rozpraszane w organizmie ludzkim, dodano regułę 21, która klasyfikuje takie wyroby odpowiednio do klas IIa, IIb lub III. Większość wyrobów substancyjnych obecnych na rynku będzie wymagała udziału jednostki notyfikowanej w ocenie zgodności.
- weryfikacja dowodów klinicznych jakimi dysponujemy dla naszego wyrobu wraz z analizą czy dane kliniczne opierają się na wykazaniu równoważności z komparatorem. Jeżeli tak, koniecznie zapoznaj się z dokumentem MDCG 2020-5, w którym Grupa Koordynacyjna ds. Wyrobów Medycznych przedstawiła nowe zasady wykazywania równoważności.
Rozwój nowych produktów
Czy jest konieczność prowadzenia badań klinicznych dla Twoich wyrobów medycznych?
Nowe rozporządzenie wprowadza zmiany w procedurach projektowania i rozwoju wyrobów. Plany projektowania i rozwoju będą musiały być dostosowane tak, aby m.in.:
- sprostać nowym wymaganiom dotyczącym dowodów klinicznych,
- formułować zalecenia dotyczące wszelkich niezbędnych dalszych obserwacji klinicznych po wprowadzeniu do obrotu,
- zapewnić spójność między planami zarządzania ryzykiem, określeniem ryzyka i korzyści, sprawozdaniami z oceny klinicznej, planami nadzoru po wprowadzeniu do obrotu i dokumentacją techniczną.
Należy dokonać przeglądu produktów znajdujących się w fazie badań i rozwoju, oraz planów ich rozwoju, aby ocenić wpływ MDR na zdolność do wprowadzenia tych produktów na rynek, a także wpływ na harmonogram prac rozwojowych.
Osoba odpowiedzialna za zgodność regulacyjną
Nowym i ważnym wymogiem MDR jest posiadanie w organizacji osoby odpowiedzialnej za zgodność
regulacyjną, o wymaganej wiedzy fachowej w dziedzinie wyrobów medycznych. Obowiązek ten dotyczy wszystkich organizacji, jednak mikro i małe przedsiębiorstwa mogą korzystać z usług takiej osoby spoza struktur własnej firmy.
Dokumentacja techniczna
Czy proces zarządzania dokumentacją umożliwia łatwe przeszukiwanie i aktualizacje przez cały cykl życia Twojego wyrobu?
Rozporządzenie MDR określa zawartość dokumentacji technicznej dla każdego wyrobu medycznego lub rodziny wyrobów medycznych. W rozporządzeniu położono większy nacisk na wymagania podejścia do zarządzania wyrobem medycznym w cyklu życia wraz z rutynową aktualizacją dokumentacji technicznej, w tym:
- w świetle informacji zebranych podczas nadzoru po wprowadzeniu do obrotu;
- wobec rozwoju stanu techniki;
- rozwoju zmian w normach lub wspólnych specyfikacjach stosowanych do wspierania oznakowania CE.
Oznakowanie produktu
Zgodnie z zasadami MDR, do każdego produktu należy załączyć informacje konieczne do zidentyfikowania wyrobu i jego producenta, a także wszelkie informacje dotyczące bezpieczeństwa stosowania oraz działań istotnych dla użytkownika wyrobu.
Informacje te mogą znajdować się na samym wyrobie, na opakowaniu lub w instrukcji użycia, a dodatkowo, jeśli producent posiada stronę internetową, powinny być na niej udostępniane i aktualizowane. Elementy, które powinny być umieszczone na etykiecie i w instrukcji używania zostały wymienione w MDR w ROZDZIALE III.
KODY UDI
Czy wiesz od kiedy wymagania dotyczące UDI będą miały zastosowanie dla Twoich wyrobów?
Kolejną nowością wprowadzoną przez MDR, związaną z oznakowaniem wyrobów medycznych, jest wdrożenie systemu UDI (Unique Device Identification). Dzięki systemowi niepowtarzalnych kodów identyfikacyjnych poprawi się zdolność rozpoznania wyrobów medycznych i skuteczność działań związanych z bezpieczeństwem po wprowadzeniu ich do obrotu.
Raporty PMS, PMCF, PSUR
Na uwagę zasługuje okres po wprowadzeniu wyrobu na rynek, w tym obszar planowania i wdrażania systemu nadzoru po wprowadzeniu do obrotu (PMS), raportowania obserwacji oraz postępowania z zewnętrznymi działaniami korygującymi dotyczącymi bezpieczeństwa. MDR wprowadza również zwiększone wymagania dotyczące planów PMS, w tym prowadzenie w razie potrzeby aktywnej obserwacji klinicznej po wprowadzeniu do obrotu (PMCF), przygotowywanie okresowych raportów o bezpieczeństwie (PSUR) dla wyrobów klasy II i klasy III, a także utrzymywanie raportów z nadzoru po wprowadzeniu do obrotu (PMSR) dostępnych dla wyrobów klasy I.
Baza EUDAMED
Nowa europejska baza danych o wyrobach medycznych – EUDAMED – będzie odgrywać istotną rolę w udostępnianiu danych o wyrobach i badaniach.
Od grudnia 2020 r. dostępny jest moduł dotyczący rejestracji podmiotów gospodarczych. Natomiast drugi moduł, dotyczący rejestracji UDI/wyrobów oraz moduł trzeci, dotyczący certyfikatów i jednostek notyfikowanych, będą dostępne do września 2021 r.
Do momentu, gdy wszystkie moduły będą dostępne, korzystanie z bazy EUDAMED jest dobrowolne. Później stanie się obowiązkiem.
Zrozumienie powyższych, wybranych, wymagań MDR dla podmiotów związanych z wyrobami medycznymi jest kluczowe dla opracowania planu wdrożenia nowych regulacji w organizacji w celu zapewnienia ciągłej zgodności z przepisami i możliwości dostarczania na rynek EU bezpiecznych wyrobów medycznych.
Przeczytaj o nowych obowiązkach dla DYSTRYBUTORÓW.
